- Visibility 19 Views
- Downloads 2 Downloads
- DOI 10.18231/j.agems.2020.013
-
CrossMark

Focusing on idiopathic pulmonary fibrosis
Introduction
The terms interstitial lung disease (ILD) and diffuse parenchymal lung disease are imprecise clinical terms for a diverse range of diseases (130 disorders) that involve inflammation and fibrosis of the alveoli, distal airways, and septal interstitium of the lungs. ILD involves abnormalities of the interstitium, the potential space between the epithelial and capillary endothelium basement membranes within the alveolus.[1] Idiopathic pulmonary fibrosis (IPF) is a common form of interstitial lung disease (ILD) and occurs predominantly in middle-aged and older adults. It accounts for 20% to 30% of ILDs and is usually progressive, resulting in respiratory failure and death. [2] The most common symptoms of IPF are dyspnea and cough. Dyspnea is usually exertional and associated with walking up inclines or steps. The cough is typically described as “dry” and “hacking,” and may start with a tickle in the throat. The severity of these symptoms varies. Other possible symptoms of IPF are fatigue and problems with sleeping. Symptoms that have not been associated with IPF include chest pain, fever, rash, weight loss, and myalgia or arthralgia, although these may be seen in various other forms of ILD. Cyanosis, cor pulmonale, peripheral oedema are observed in terminal stages. The 5-year survival rate of 20–40% associated with IPF is worse than that of cancers. Smoking, environmental exposures, gastroesophageal reflux disease (GERD), infections, and genetic predisposition are potential risk factors for the development of IPF. The incidence of IPF increases with older age, with presentation typically consisting of insidious onset of dyspnoea in the sixth and seventh decades. [3] The estimated prevalence of IPF in the United States (US) ranges from 14-43 per 100,000 population.[4] Mutations in the genes encoding telomerase (TERT and TERC) cause IPF through shortening of telomere lengths and probable exhaustion of lung stem cells. All of the mutations are individually rare, but, collectively, TERT mutations are the most common genetic defect found in Familial pulmonary fibrosis. The overall penetrance of pulmonary fibrosis in TERT mutation carriers is 40% in subjects with a mean age of 51 years. Penetrance increases with advanced age, and is positively associated with fibrogenic environmental exposures. [5] IPF is associated with pulmonary or extrapulmonary comorbidities. Pulmonary comorbidities include pulmonary hypertension, emphysema, and lung cancer, while non-pulmonary conditions include venous thromboembolism, coronary artery disease, congestive heart failure, sleep-disordered breathing, gastro-oesophageal reflux disease, and anxiety or depression. [6]
Pathogenesis of IPF

In an initiating phase, there is lung alveolar epithelial damage with loss of the normal lung architecture and disruption of the basement membrane across which gas exchange takes place. With further epithelial damage and apoptosis, comes upregulation of epithelial integrins, such as αvβ6, and a phase of fibroproliferative repair dominates – driven by high levels of TGF-β. Locally activated TGF-β drives the recruitment of fibroblasts and a feed-forward cycle of further TGF-β production. Under these conditions, fibroblasts differentiate into myofibroblasts that express high levels of integrin αvβ6, are resistant to apoptosis and lay down a collagen matrix. Once collagen has been laid down in a lung, the architecture of which is already distorted, gas exchange is no longer efficient. [7]
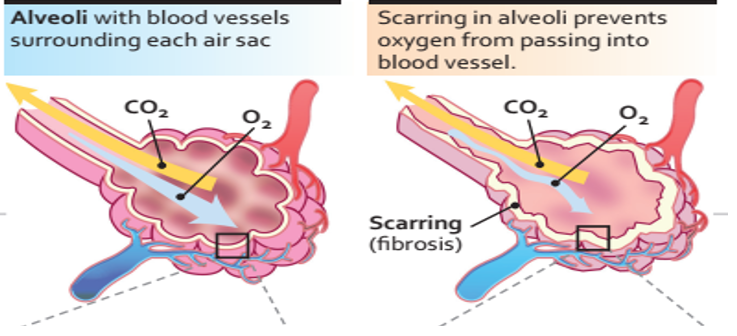
Diagnosis of IPF
IPF should be considered in middle-aged/elderly individuals with unexplained chronic exertional dyspnea. It commonly presents with cough and bibasilar inspiratory crackles (sometimes referred to as “Velcro rales” because of their sound) and less often with finger clubbing (spoon-shaped nails). Spirometry may be normal or show restriction (in contrast to obstruction such as in chronic obstructive pulmonary disease [COPD]). Diagnosis of IPF requires exclusion of known causes of ILD, including systemic autoimmune diseases and environmental exposures, and the presence of a usual interstitial pneumonia (UIP) pattern on high-resolution computed tomography or lung biopsy. HRCT features frequently seen in UIP include honeycombing, traction bronchiectasis, and traction bronchiolectasis, which may be seen with the concurrent presence of ground-glass opacification and fine reticulation. [8]
Drugs causing pulmonary fibrosis
Now there are over 450 drugs recognizing as being implicated in interstitial lung disease.
Antibiotics, particularly nitrofurantoin
Immunosuppressant drugs, such as methotrexate
The mortality in acute amiodarone pulmonary toxicity approaches 40-50%.
Cancer chemotherapy drugs Any chemotherapeutic drug can adversely affect the lung, but the drugs most commonly implicated in lung toxicity are bleomycin, carmustine, busulfan, and cyclophosphamide . Approximately 1-10% of patients taking one of these drugs are affected Classes of biological agents with reported DILD include tumor necrosis factor (TNF)-alfa blockers, anti-CD20 antibodies, recombinant interferon (INF) alpha, T-cell antiproliferative agents, or others like cetuximab, bevacizumab, alemtuzumab or trastuzumab. Hazardous associations have been reported with the coadministration of cisplatin and bleomycin, which can increase the risk of bleomycin induced interstitial lung disease. The combination therapy of gemcitabine and bleomycin is very toxic. [9], [10]
Managing devastating disorder
Regardless of the cause of IPF the goals of treatment are:
To decrease inflammation and prevent further lung scarring
To remove the source of the problem, whenever possible
To minimize and manage potential complications
Pharmacological Treatment
The treatment approach to IPF has evolved considerably over the last two decades. A typical regimen in the year 2000 involved immunosuppression with prednisolone and azathioprine. A combination of prednisone, azathioprine, and N-acetylcysteine (NAC) has been widely used as a treatment for idiopathic pulmonary fibrosis. N-Acetylcysteine (NAC) at a dose of 600mg three times daily, added to prednisolone preserves Forced Vital Capacity (FVC) and Carbon Monoxide Diffusing Capacity (DLCO), thus slowing the deterioration of pulmonary function in patients with Idiopathic Pulmonary Fibrosis (IPF) better than prednisolone mono therapy. [11]
Prednisone is used to treat and prevent inflammation by inhibiting the immune system. While prednisone is not usually used to treat idiopathic pulmonary fibrosis, it is sometimes used to treat inflammation in the lungs of people living with other forms of pulmonary fibrosis. Since prednisone suppresses the immune system, it can potentially increase the frequency and severity of infections.
Azathioprine is prescribed as part of the treatment of a number of ILDs (not IPF), in combination with prednisolone as a steroid sparing agent attempting to halt the progression of fibrotic disease. In most ILD patients, azathioprine will be trialed for a minimum period of three months (British Thoracic Society Interstitial Lung disease guideline, September 2008).
Therapy with acetylcysteine at a dose of 600 mg three times daily, added to prednisone and azathioprine, preserves vital capacity and DLCO (carbon monoxide diffusing capacity) in patients with idiopathic pulmonary fibrosis better than does standard therapy alone. [12]
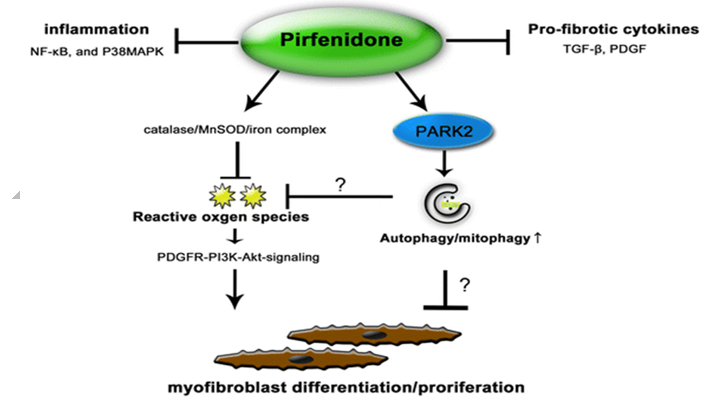
Pirfenidone is used for the treatment of idiopathic pulmonary fibrosis. It works by reducing lung fibrosis through downregulation of the production of growth factors and procollagens I and II. The first Phase III clinical trial to evaluate the efficacy and safety of pirfenidone for the treatment of patients with idiopathic pulmonary fibrosis was conducted in Japan. This was a multicentre, randomised, double-blind, trial, in which 275 patients with idiopathic pulmonary fibrosis were randomly assigned to receive pirfenidone 1800 mg/day (110 patients), pirfenidone 1200 mg/day (56 patients), or placebo (109 patients), for 52 weeks. Pirfenidone 1800 or 1200 mg/day reduced the mean decline in vital capacity from baseline to week 52 compared with placebo. Progression-free survival was also improved with pirfenidone compared with placebo.[13]
Pirfenidone targets: Different targets in vivo and in vitro for PFD have been described, the most prominent being inhibition of TGF-β1 and TNF-α. However, it has also been shown that Pirfenidone has either direct or indirect action on other molecules such as collagen I, PDGF, IL-6, IL-1β, IL-13, IL-12p40, fibronectin, HSP47 and ICAM-1. To be taken with food. Recommended dosage: 801 mg three times daily (2403 mg/day). The most common adverse reactions (≥10%) are nausea, rash, abdominal pain, upper respiratory tract infection, diarrhea, fatigue, headache, dyspepsia, dizziness, vomiting, anorexia, gastro-esophageal reflux disease, sinusitis, insomnia, weight decreased, and arthralgia.[14], [15]
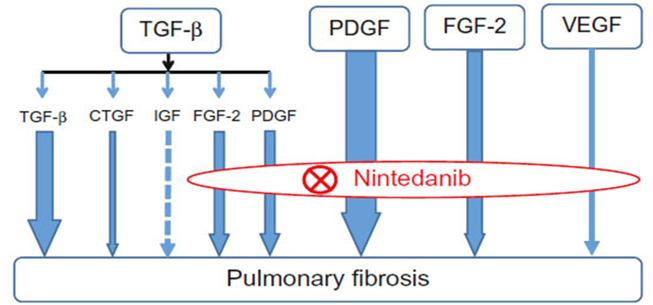
Nintedanib: binds to the intracellular ATP binding pocket of fibroblast growth factor receptors (FGFRs), platelet-derived growth factor receptors (PDGFRs) and vascular endothelial growth factor receptors (VEGFRs) resulting in blockage of the autophosphorylation of these receptors and the downstream signalling cascades.
Nintedanib inhibited TGF-β-induced (10ng.ml/l) fibroblast to myofibroblast transformation of primary human lung fibroblasts from IPF patients, as determined by α-smooth muscle actin mRNA expression as a marker for myofibroblast differentiation, with an estimated IC50 value of 144 mmol/l.
Recommended dosage: 150 mg twice daily approximately 12 hours apart taken with food, Most common adverse reactions (≥5%) are: diarrhea, nausea, abdominal pain, vomiting, liver enzyme elevation, decreased appetite, headache, weight decreased, and hypertension.
Treatment with nintedanib may slow decline in lung function, decrease the frequency of exacerbations, and improve quality of life in subjects with IPF. High-dose nintedanib improved the quality of life, slowed the progression of lung fibrosis and the decline of lung function, and reduced the rate of exacerbations in individuals with mild and moderate IPF.[16], [17]
Rituximab works by destroying B cells, a type of white blood cell, called a B-lymphocyte, which produce autoantibodies. Rituximab represents the most widely used biologic treatment in patients with rapidly progressive RA-ILD who are unresponsive to first line therapeutic compounds including corticosteroids and methotrexate In IPF research study, rituximab will be given into a vein to reduce the autoantibody levels that believe might be contributing to the lung damage in IPF.[18]
Domiciliary oxygen therapy is often prescribed for patients with hypoxaemia due toadvanced lung disease, most commonly chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD). Long-term oxygen therapy (LTOT) trials conducted in patients with COPD in the 1980s remain the basis for clinical decisions and guideline recommendations regarding LTOT for patients with non-COPD conditions as there is a lack of high-quality evidence concerning its use in the non COPD population.[19]
Non Pharmacologic treatment
The majority of non-pharmacological management strategies apply to all patients with fibrotic ILD, regardless of progression or the underlying etiology. Smoking cessation, antigen avoidance, protection from occupational exposures, and cessation of medications that can potentially cause ILD are all important. Pneumococcal vaccination and annual influenza vaccination are also appropriate in almost all patients with fibrotic ILD. [20]
Developing Targets
Galectin-3: is another circulating protein which plays a key role in fibrosis development through the activation of macrophages and myofibroblasts . (Galecto Biotech AB, Copenhagen, Denmark, has been developed as a specific inhibitor of the galactoside binding pocket of galectin-3. It is administered via inhalation and is currently being investigated in a phase I trial.
Pentraxin-2 (PTX-2), also known as serum amyloid P, is a circulating protein which binds to Fc-gamma receptors on monocytes and promotes epithelial healing. Its being evaluated is in Phase 1 study.
(IL-13) is a T-helper type 2 (Th2) cell cytokine that promotes lung fibrogenesis in a number of experimental settings. Lebrikizumab, tralokinumab are under evaluation.
A Phase 2 open label trial of Pamrevlumab a monoclonal antibody blocking the downstream effects of connective tissue growth factor (CTGF), showed an acceptable safety and efficacy profile and thus a phase III clinical trial is currently anticipated.[21], [22]
Conclusion
IPF mostly afflicts people 50–70 years of age, The prognosis is poor, with a mean survival of about 2.5–5 years after definite diagnosis which makes IPF as a non benign disease which is difficult to predict. At present, nintedanib and pirfenidone {Anti-fibrotic agents} are routinely used to treat IPF based on phase III clinical trials demonstrating a reduced rate of decline in FVC over time among patients with IPF taking either medication. Definitive long-term therapy is lung transplantation where also the results are not very encouraging.
Conflict of Interest
None.
Source of Funding
None.
References
- S J Bourke. Interstitial lung disease: progress and problems. Postgraduate Med J 2006. [Google Scholar] [Crossref]
- JH Ryu, T Moua, C E Daniels, T E Hartman, E S Yi, JP Utz. Idiopathic Pulmonary Fibrosis: Evolving Concepts. Mayo Clin Proce 2014. [Google Scholar] [Crossref]
- L. Nalysnyk, J. Cid-Ruzafa, P. Rotella, D. Esser. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012. [Google Scholar] [Crossref]
- . www,rarediseasesindia,org/pulmonaryfibrosis. . [Google Scholar]
- C K Garcia. Idiopathic pulmonary fibrosis: update on genetic discoveries. Proc Am Thorac Soc 2011. [Google Scholar]
- P R Ortega, M Molina-Molina. Interstitial Lung Diseases in Developing Countries. Ann Global Health 2019. [Google Scholar] [Crossref]
- T A Mikolasch, H S Garthwaite, J C Porter. Update in diagnosis and management of interstitial lung disease. Clin Med 2017. [Google Scholar] [Crossref]
- R Pleasants, R M Tighe. Management of Idiopathic Pulmonary Fibrosis. Ann Pharmacoth 2019. [Google Scholar]
- R Rafii, M M J Timothy, E Albertson1, L Andrew. ChanA review of current and novel therapies for idiopathic pulmonary fibrosis. J Thoracic 2013. [Google Scholar] [Crossref]
- M Schwaiblmair, W Behr, T Haeckel, B Märkl, W Foerg, T Berghaus. Drug Induced Interstitial Lung Disease. Open Respir Med J 2012. [Google Scholar] [Crossref]
- . The Idiopathic Pulmonary Fibrosis Clinical Research NetworkPrednisone, Azathioprine, and N-Acetylcysteine for Pulmonary Fibrosis. N Engl J Med 2012. [Google Scholar]
- M Demedts. High-Dose Acetylcysteine in Idiopathic Pulmonary Fibrosis. N Engl J Med 2005. [Google Scholar]
- H. Taniguchi, M. Ebina, Y. Kondoh, T. Ogura, A. Azuma, M. Suga, . Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2010. [Google Scholar] [Crossref]
- G Margaritopoulos, E Vasarmidi, K Antoniou. Pirfenidone in the treatment of idiopathic pulmonary fibrosis: an evidence-based review of its place in therapy. Core Evid 2016. [Google Scholar] [Crossref]
- D Alejandro. Role and New Insights of Pirfenidone in Fibrotic Diseases. Int J Med Sci 2015. [Google Scholar]
- E L Wollin, A Wex, G Pautsch, K E Schnapp, S Hostettler, Stowasser. Martin KolbMode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 2015. [Google Scholar]
- I. A. Dimitroulis. Nintedanib: A Novel Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Respiratory Care 2014. [Google Scholar] [Crossref]
- J Gregory, Toby M Keir, Maher. Rituximab in severe, treatment-refractory interstitial lung disease. Respirol 2014. [Google Scholar]
- H Yet. Khor 1,2,3,4, Elisabetta A. Renzoni5, Dina Visca 6,7, Christine F. McDonald1,2,4 and Nicole S. L. Goh Oxygen therapy in COPD and interstitial lung disease: navigating the knowns and unknowns. ERJ Open Res 2019. [Google Scholar]
- AW Wong, C J Ryerson, S A Guler. Progression of fibrosing interstitial lung disease. Respir Res 2020. [Google Scholar] [Crossref]
- S Barratt, A Creamer, C Hayton, N Chaudhuri. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J Clin Med 2018. [Google Scholar] [Crossref]
- T Karampitsakos1, A Vraka. Demosthenes Bouros2, Stamatis-Nick Liossis3 and Argyris Tzouvelekis, Biological treatments of interstitial ling diseases. Front Med 2019. [Google Scholar]